Myopathies mitochondriales
Les myopathies mitochondriales affectent les muscles : intolérance à l’effort, faiblesse musculaire, fatigue, douleur… D’autres organes sont souvent touchés (cerveau, yeux…). Elles nécessitent un suivi pluridisciplinaire au sein de consultations spécialisées. Les recherches en cours visent à mieux comprendre ces maladies très complexes et parvenir à les traiter.
Que sont les myopathies mitochondriales ?
Ce sont des maladies génétiques rares dues au mauvais fonctionnement des mitochondries, le fournisseur énergétique de nos cellules. Elles représentent un ensemble de maladies qui diffèrent, parfois beaucoup, les unes des autres. Elles comprennent notamment le syndrome de MELAS, le syndrome de MERRF, le syndrome de Kearns-Sayre, l'ophtalmoplégie progressive…
Toutes formes confondues les myopathies mitochondriales toucheraient une personne sur 4 300.
Toutes les maladies mitochondriales ne sont pas des myopathies, c’est-à-dire causées par une atteinte des cellules musculaires. Ainsi, dans le syndrome de Leigh qui fait partie des maladies mitochondriales les plus communes, ce sont les cellules du système nerveux central (tronc cérébral et cerveau) qui sont atteintes en premier, entrainant des difficultés visuelles et respiratoires ainsi que motrices (manque de contrôle et de coordination des mouvements).

Quand les cellules manquent d’énergie
Chaque cellule possède des mitochondries qui lui fournissent l’énergie nécessaire à son fonctionnement. Leur nombre s’accroit dans les organes à fort besoin en énergie (muscle, cerveau, nerf…).
Dans les cellules musculaires, plusieurs milliers de mitochondries sont présentes le long des fibres musculaires pour fournir l’énergie nécessaire au travail musculaire.
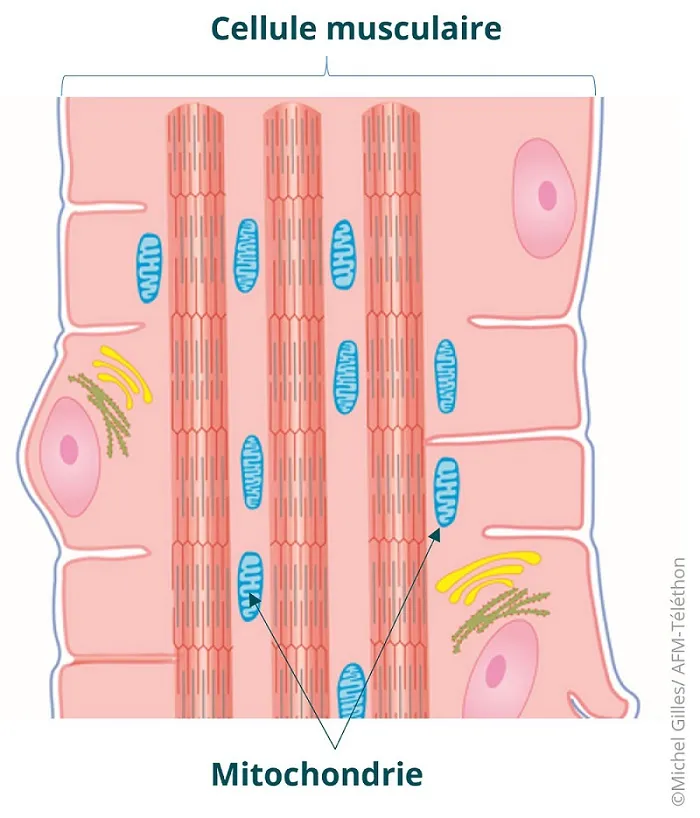
Dans les myopathies mitochondriales, les mitochondries fonctionnent mal et les cellules musculaires n’ont pas l’énergie dont elles ont besoin, ce qui entraine une fatigabilité qui peut évoluer vers une fonte et une faiblesse musculaire si des cellules dégénèrent.
D’autres organes peuvent aussi être touchés (on parle de maladie « multisystémique »), principalement le cerveau (encéphalopathie) et le cœur (cardiomyopathie, troubles de la conduction) dont les besoins énergétiques sont très importants mais aussi les yeux (atteinte de la rétine, atrophie du nerf optique), les oreilles (surdité), les reins, le foie ou l’appareil digestif (difficultés à avaler, diarrhée ou constipation...).
Que peut-on faire ?
Les symptômes et l’évolution des myopathies mitochondriales dépendent de la forme de myopathie mitochondriale : fatigue, manque de tonus et de force musculaires, mauvais équilibre, douleurs, difficultés visuelles ou auditives, atteinte cardiaque, épisode ressemblant à un accident vasculaire cérébral, crises d’épilepsie… Chez certaines personnes, les manifestations sont si légères qu’elles peuvent passer longtemps inaperçues alors que chez d’autres, elles sont très sévères dès la naissance.
La prise en charge est adaptée au cas par cas et comprend :
- la rééducation, la balnéothérapie et les massages, pour entretenir les muscles.
- la kinésithérapie, le port d’attelles et parfois la chirurgie, pour limiter les rétractions musculaires et maintenir la souplesse des articulations.
- des séances de kinésithérapie respiratoire, parfois une ventilation non invasive nocturne, en cas d’atteinte respiratoire.
- un traitement par coenzyme Q10 (à la fois fournisseur d’énergie et antioxydant) est parfois proposé (en cas de déficit en CoQ10) et peut s’avérer efficace.
- le conseil génétique, pour informer et accompagner une personne ou une famille confrontée au risque de développer ou de transmettre la maladie.
- une prise en charge spécifique des atteintes associées : médicaments contre les crises d’épilepsie ou le diabète, médicaments et/ou pacemaker pour les manifestations cardiaques, appareil auditif pour une surdité, lunettes spéciales ou chirurgie pour la chute des paupières…
Vers une prise en charge adaptée pour les malades à risque d’une atteinte cardiaque
Les personnes atteintes de maladies mitochondriales sont à risque de développer une insuffisance cardiaque et/ou des troubles du rythme cardiaque (arythmie). Une étude internationale financée par l’AFM-Téléthon a mis en évidence un lien entre la survenue d’une atteinte cardiaque, et le type d’anomalie génétique.
L’identification des anomalies génétiques en complément du suivi cardiaque pour dépister une maladie du cœur dès les premiers signes permettront d’estimer les risques de développer une atteinte cardiaque grave et donc assurer une prise en charge adaptée préventive chez les malades les plus à risque.
Où consulter ?
Un réseau expert de consultations dans toute la France
La prise en charge d’une personne atteinte de myopathie mitochondriale fait intervenir des professionnels médicaux et paramédicaux : neurologue, médecin de rééducation, généticien, ophtalmologiste, kinésithérapeute…) et qui travaillent en collaboration avec d’autres professionnels de santé (gynécologue-obstétricien, endocrinologue, diabétologue, interniste...).
Les consultations pluridisciplinaires spécialisées dans les maladies neuromusculaires offrent l’avantage de regrouper au même endroit ces spécialités médicales. Elles sont structurées en une filière de santé des maladies rares neuromusculaires (FILNEMUS). Réparties sur tout le territoire, ces consultations peuvent assurer le diagnostic d’une myopathie mitochondriale, la prise en charge et le suivi dans la durée à proximité du domicile des personnes atteintes.
Deux Centres de référence nationaux
Spécialisés dans les maladies mitochondriales, ces centres qui appartiennent au réseau FILNEMUS assurent à la fois un suivi médical régulier et organisent la prise en charge de proximité.
• le Centre de Référence pour les Maladies Mitochondriales de l’Enfant à l’Adulte (CARAMMEL) réunit les CHU d’Angers, Bordeaux, Bicêtre (Kremlin-Bicêtre), Necker et Lariboisière (Paris). Il est associé aux centres de compétence des CHU de Dijon, Limoges, Rouen et Tours.
• le Centre de référence des maladies mitochondriales Sud-Méditerranée CALISSON associe les CHU de Marseille et de Nice.
Comment se transmettent-elles ?
Les composants de mitochondries sont fabriqués à partir de l’information génétique, l’ADN, localisé :
- soit au niveau des chromosomes du noyau, l’ADN nucléaire,
- soit au niveau de la mitochondrie elle-même, l’ADN mitochondrial.
Des anomalies de ces gènes, qui interviennent dans le fonctionnement des mitochondries sont à l’origine des myopathies mitochondriales. Plus de 350 gènes responsables de maladies mitochondriales ont été identifiés.
- Lorsque l’anomalie génétique touche l’ADN nucléaire, la maladie peut se transmettre à la descendance, selon différents modes de transmission génétiques (autosomique dominant ou récessif, lié à l’X).
- Les anomalies de l'ADN mitochondrial ont une transmission dite « mitochondriale » (ou maternelle) : seul l’ADN mitochondrial de la mère est transmis à l’enfant car les mitochondries des spermatozoïdes ne participant pas à la formation de l’embryon lors de la fécondation.
L’hétéroplasmie
Lorsqu’une anomalie génétique est présente au niveau de l’ADN mitochondrial, elle n’est généralement pas présente sur l’ensemble des mitochondries de chaque cellule : certaines mitochondries sont affectées, d’autres non. La proportion entre les deux varie d’une cellule à l’autre et au cours du temps. On parle d’hétéroplasmie.
Comment sont-elles diagnostiquées ?
Le diagnostic d’une myopathie mitochondriale est souvent une démarche longue : ces maladies sont rares et complexes, les symptômes peuvent varier d’une personne à l’autre et plus de 350 gènes ont été identifiés, compliquant l’analyse génétique.
Les symptômes (et notamment l’association d’une atteinte des muscles à celle d’autres organes), l’examen clinique, l’électromyogramme et la biopsie musculaire permettent au médecin d’évoquer le diagnostic de myopathie mitochondriale. Un bilan plus général (cœur, yeux, cerveau…) fournit des arguments en faveur d’une maladie des mitochondries et peut aider à préciser son type. Des analyses spécialisées (études de la chaîne respiratoire, épreuve d’effort…) y contribuent également.
Néanmoins, seules les études génétiques, à partir d’une prise de sang ou d’un prélèvement de muscle, permettent de poser le diagnostic avec certitude. Parfois, il n’est pas possible d’identifier d’anomalies génétiques sur les gènes connus alors que les symptômes sont très évocateurs.
Un meilleur diagnostic génétique des myopathies mitochondriales
Avec l’amélioration des méthodes de diagnostic moléculaire, comme les techniques de séquençage de nouvelle génération (NGS) qui permettent d’analyser simultanément des centaines de gènes, voire l’ensemble des gènes d’une cellule, le diagnostic génétique des myopathies mitochondriales s’est considérablement amélioré ces dernières années.
Un grand nombre de nouvelles anomalies génétiques en cause, tant au niveau de l’ADN mitochondrial que de l’ADN nucléaire, ont ainsi été identifiées, tels que celles des gènes MT-CO3 ou COX6A2.
Les connaissances sur les anomalies génétiques mitochondriales et les techniques pour les analyser sont partagées et rassemblées grâce à des bases de données et des articles médicaux. Cela permet, petit à petit, de faciliter le diagnostic génétique de ces maladies, de mieux comprendre les mécanismes en cause et de soutenir le développement de nouvelles stratégies thérapeutiques.
Le réseau MitoDiag
Le réseau MitoDiag, créé en 2000, regroupe l’ensemble des laboratoires français impliqués dans le diagnostic des maladies mitochondriales. Ces laboratoires participent à une base de données Mitomatcher qui recueille les données cliniques et biologiques de plus de 3 000 personnes atteintes de myopathies mitochondriales.
Lutter contre l’errance diagnostique grâce au projet COMMI
Le réseau MITODIAG a annoncé en mai 2023 le lancement du projet COMMI pour constituer une cohorte regroupant 400 patients atteints de maladie mitochondriale liée à une ou plusieurs mutations sur l’ADN nucléaire.
L’analyse de leurs données cliniques et génétiques permettra de mieux définir les liens entre une anomalie génétique et ses manifestations de la maladie. L’objectif est de simplifier l’interprétation des résultats d’analyses génétiques et, au-delà, de faire progresser la prise en charge, l’accès aux essais cliniques et les stratégies de diagnostic.
Une FIV pour remplacer les mitochondries
Dans le cas des maladies liées à des gènes mitochondriaux, des techniques de fécondation in vitro (FIV) avec remplacement mitochondrial ont été mises au point pour prévenir la transmission d’une anomalie de l’ADN mitochondrial de la mère à son enfant, comme dans certaines myopathies mitochondriales.
Le noyau d’un ovule d’une mère atteinte de myopathie mitochondriale est prélevé puis transféré dans un ovule d’une femme donneuse indemne de la maladie dont on avait préalablement enlevé le noyau. Ainsi, cet ovule possède le noyau de la mère et les mitochondries sans anomalie génétique de la donneuse. Puis la fécondation est réalisée in vitro avec le spermatozoïde du père (on parle parfois de FIV à trois parents ou trois ADN).
Cette technique de fécondation in vitro avec remplacement mitochondrial a permis la naissance de premiers bébés indemne de myopathie mitochondriale. Encore controversée et suscitant des questionnements éthiques, elle est interdite en France et aux États-Unis notamment, mais autorisée au Royaume-Uni.
Des traitements à l’étude
Ces dernières années, plusieurs traitements ont été testés dans les myopathies mitochondriales, certains en laboratoire dans des modèles cellulaires ou animaux, d’autres déjà à l’essai chez l’homme. Ils visent à améliorer certaines manifestations de ces maladies.
- L’élamiprétide (ou MTP-131) est un candidat-médicament évalué dans plusieurs essais cliniques successifs. Cette molécule, qui pénètre dans les mitochondries augmente la production d’énergie : elle a permis d’améliorer la performance au test de marche de 6 minutes en particulier chez les participants présentant des anomalies de l’ADN nucléaire et de réduire leur fatigue. L’essai NuPower évalue son efficacité et sa sécurité d’utilisation chez 130 malades avec des mutations nucléaires pendant un an dans une dizaine de pays.
- Une supplémentation en niacine (ou vitamine B3), laquelle favorise la production de NAD (pour nicotinamide adénine dinucléotide), une molécule manquante dans certaines myopathies mitochondriales, améliore la force musculaire des malades.
- La pratique d’un entrainement physique aérobie régulier (c’est-à-dire à faible puissance pendant une longue durée) a montré une tendance à augmenter le nombre de mitochondries dans les muscles, réduire la fatigue et améliorer la qualité de vie des malades.
- Des régimes cétogènes, c’est-à-dire riches en graisses et pauvres en sucres, stimulent le développement des mitochondries et semblent améliorer la force et la régénération musculaires des malades.
Dans le déficit en TK2
- Un traitement à base de nucléosides, qui entrent dans la composition de l’ADN et de l’ARN, a montré des effets bénéfiques sur les fonctions motrices et respiratoire de personnes atteintes de myopathie mitochondriales avec déficit en TK2.
Dans le syndrome de MELAS
- Une supplémentation en L-arginine, dont la quantité est réduite dans le syndrome de MELAS, diminue la fréquence et la sévérité des accidents vasculaires cérébraux dans cette maladie. L’innocuité et l’efficacité d’autres suppléments (glutamine, L-citrulline…) et molécules (dichloroacétate, sonlicromanol, vatiquinone…) sont également testées dans plusieurs essais cliniques.
- La vatiquinone (EPI-743, PTC-743) a pour objectif de combler le déficit en glutathion dans les maladies mitochondriales. Plusieurs études réalisées chez des patients atteints d’un syndrome de Leigh traités avec cette molécule ont déjà montré une amélioration de leurs symptômes. La vatiquinone a obtenu en 2022 la désignation de médicament orphelin en Europe afin de favoriser son développement. Un essai est en cours dans sept pays dont la France pour évaluer son effet dans plusieurs maladies mitochondriales, dont les syndromes de Leigh et de MELAS.
- La N-Acétylcystéine (NAC) est un antioxydant qui permet de stimuler la production de glutathion. Cet élément est souvent en quantité insuffisante chez les patients atteints de MELAS, réduisant les capacités du cerveau à réparer certains dommages dus au stress oxydatif. Un essai est en cours chez 18 adultes malades pour évaluer l’innocuité d’une prise de NAC pendant trois mois, et ses effets sur les niveaux de glutathion dans le cerveau, les capacités cognitives et motrices, et la qualité de vie. Sur le même principe, un essai est en cours pour évaluer l’efficacité du TTI-0102 dont le composé actif est la cystéamine, un autre antioxydant précurseur du glutathion.
- Le zagociguat, un modulateur de l’activité de la guanylate cyclase, une enzyme impliquée dans la plasticité synaptique, est à l’étude dans l’essai PRIZM chez 44 malades afin de déterminer notamment son efficacité sur l’amélioration de la fatigue et des performances cognitives.
Identifier de nouveaux candidats-médicaments
L’AFM-Téléthon finance depuis plusieurs années une collaboration entre des équipes françaises spécialisées dans les maladies mitochondriales, dont l’équipe Mitolab dirigée par le Pr Vincent Procaccio (à Angers), à l’origine de deux programmes de recherche :
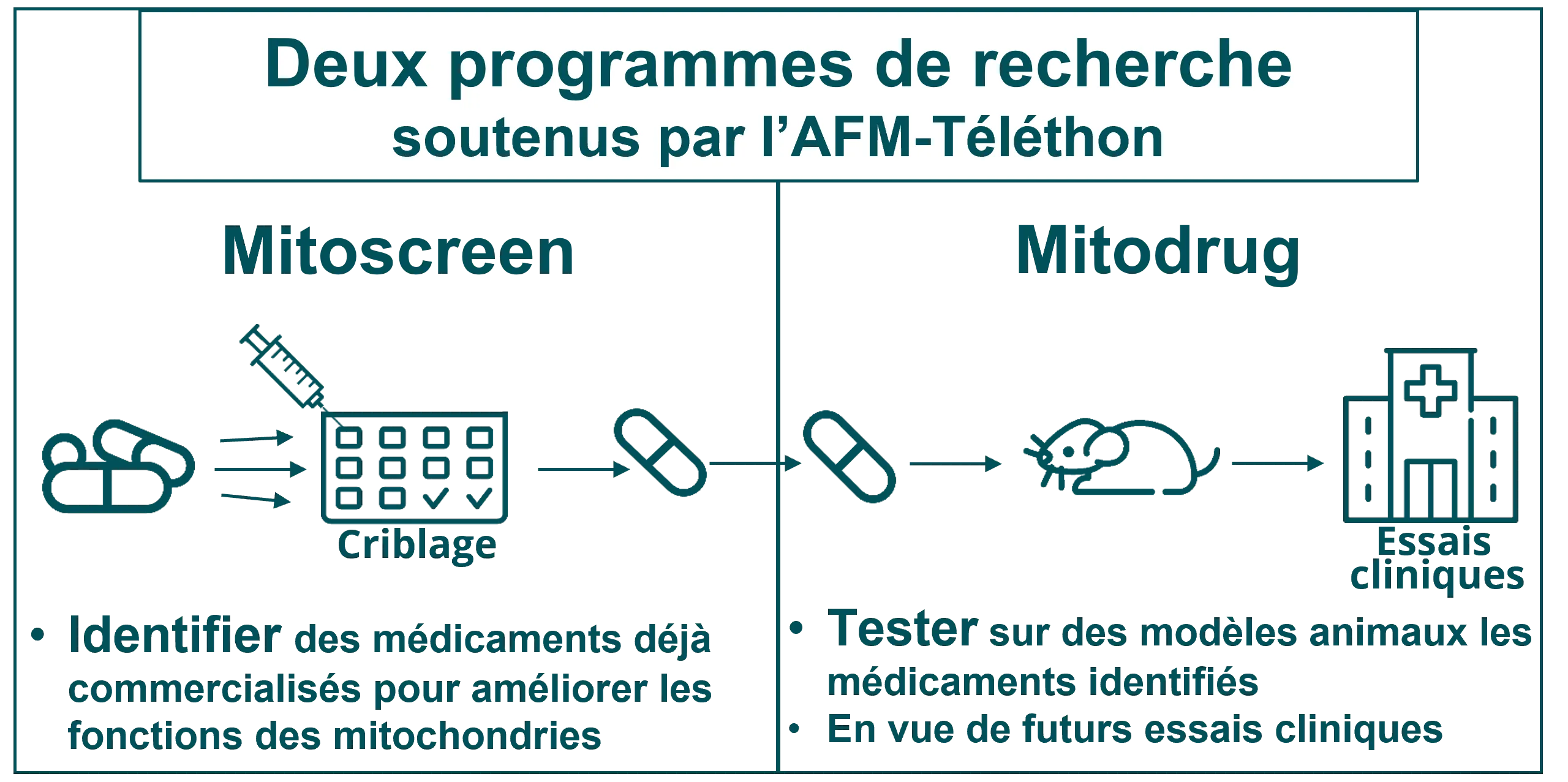
Mitoscreen, pour identifier de nouveaux traitements
Ce projet a permis d’identifier, par criblage moléculaire, plusieurs médicaments déjà utilisés dans le traitement d’autres maladies et capables d’améliorer les fonctions des mitochondries dans des modèles cellulaires de maladie mitochondriale. Ce « repositionnement thérapeutique » permet d’accélérer le lancement d’essais cliniques, les études de tolérance et de toxicologie ayant déjà été réalisées pour ces produits.
Mitodrug, pour tester ces nouveaux traitements
Le programme Mitodrug a pris le relais : il teste, dans des modèles de souris, cinq médicaments identifiés dans le cadre de Mitoscreen et vise à comprendre leur mécanisme d’action en vue de préparer de futurs essais chez l’homme.
La thérapie génique
Des approches de thérapie sont à l'étude dans plusieurs myopathies mitochondriales dont l’encéphalopathie myo-neuro-gastrointestinale (MNGIE), le déficit en TK2 et le syndrome de MELAS. Les premiers résultats sur des souris malades sont encourageants sur la faisabilité de cette piste thérapeutique.
Des congrès internationaux
Le congrès « Myology-MitoNice2022 » s'est déroulé à Nice du 12 au 17 septembre 2022 : il associait MitoNice, un congrès international dédié à la médecine mitochondriale et le 7ème Congrès International de Myologie, organisé par l’AFM-Téléthon.
D’autres congrès, dédiés spécifiquement aux myopathies mitochondriales, ou portant sur les maladies neuromusculaires au cours desquels la recherche sur les myopathies mitochondriales est régulièrement présentée comme le congrès de la World Muscle Society, sont l’occasion de réunir des chercheurs impliqués dans ces maladies.
MeetOchondrie, un réseau devenu association
En France, de 2006 à 2015, le réseau « MeetOchondrie » a rassemblé les acteurs de la recherche fondamentale, clinique ou appliquée dans le domaine des mitochondries. Dans la continuité de ce réseau, l’association « Réseau MeetOchondrie » a été créée en 2015 afin de fédérer les chercheurs travaillant sur les mitochondries, notamment en recherche fondamentale, physiologique et pathologique. Elle bénéficie de la participation de différentes associations impliquées dans les maladies mitochondriales comme l’AFM-Téléthon.
Elle organise régulièrement des colloques ou des webinaires.



Comment se manifestent-elles ?
Des signes différents d’une maladie à l’autre
Une évolution très variable d’une forme à l’autre et d’une personne à l’autre
La plupart des myopathies mitochondriales sont lentement évolutives Elles se caractérisent souvent par des périodes d’exacerbation, souvent provoquées par un effort plus important que d’habitude, une fièvre ou un stress.
En l’absence de prise en charge adaptée, l’atteinte des muscles peut conduire à des complications orthopédiques (rétractions musculo-tendineuses, limitation de l’amplitude articulaire…) et respiratoires.
Chez le nourrisson, il existe à la fois des formes graves et des formes de meilleur pronostic. Ainsi, il existe de très rares cas de myopathie mitochondriale dite « bénigne du nourrisson » qui guérit de façon spontanée.