
Les essais cliniques en pratique
Participer à un essai clinique est une démarche individuelle pour un bénéfice collectif : devenir l’un des acteurs de l’évaluation d’un futur traitement qui pourrait servir à un plus grand nombre de malades. Mais entre espoir et inquiétude, cette décision doit être prise après mûre réflexion et en ayant reçu une information la plus complète possible.
Qu’est-ce qu’un essai clinique ?
Un essai clinique (ou thérapeutique) consiste à évaluer chez l’Homme les effets d’un traitement potentiel (un candidat-médicament, un dispositif médical…) dans une maladie afin de s’assurer qu’il est bien toléré et efficace. C’est un préalable incontournable à la mise à disposition d’un nouveau traitement sur le marché.
Une étude « interventionnelle »…
Une étude clinique est qualifiée d’interventionnelle quand elle implique une intervention non dénuée de risque pour les personnes qui y participent, et non justifiée par leur prise en charge habituelle. Il s’agit notamment d’évaluer les effets d’un traitement, auprès du malade, dans des conditions strictement contrôlées.
Elle se différencie ainsi d’une étude non-interventionnelle ou d’une étude observationnelle laquelle suit les malades, sans intervenir, pour mieux décrire et connaître l’histoire naturelle des maladies, identifier de meilleurs outils diagnostiques ou de suivi...
…qui fait suite à des études précliniques
Ce n'est qu'une fois que les études précliniques ont apporté la preuve que le traitement est susceptible d’être efficace et bien toléré chez l’Homme que les autorités réglementaires, sur la base de ces résultats précliniques, peuvent répondre favorablement à la demande d’autorisation d’essai clinique.
En quoi consiste la recherche préclinique ?
La recherche préclinique correspond à l’étude du comportement de candidats-médicaments dans des cellules en culture (in vitro) et des modèles animaux (in vivo), étapes préalables indispensables à l’administration d’un candidat-médicament chez l’homme. Au cours de cette phase préclinique, les chercheurs étudient la pharmacologie, la pharmacocinétique et la toxicologie de la molécule : mécanismes d’action, propriétés physico-chimiques, devenir du composé dans l’organisme, organes ciblés, toxicité… La recherche préclinique permet ainsi de déterminer une première estimation de la dose, sans effet toxique, que l’on pourra administrer chez l’homme. Ces données sont indispensables à la constitution du dossier de demande d’autorisation de mise sur le marché (AMM) du futur médicament auprès des agences règlementaires.
Un développement clinique en quatre phases
Pour obtenir des preuves fiables de la sécurité et de l’efficacité de son utilisation chez l’Homme, le traitement est testé au cours d’essais successifs, correspondant à différentes phases : I, II, III, IV. Chaque phase permet de fournir des réponses spécifiques sur le produit testé : est-ce que le produit est bien toléré ? Quelle est la dose optimale ? Quelle est son efficacité ?...
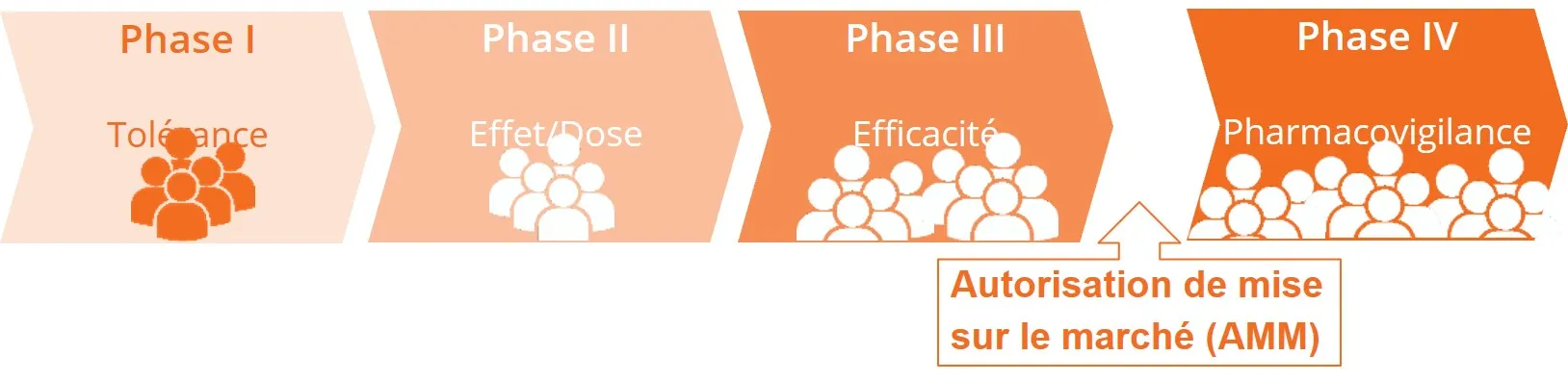
Essai de phase I : le produit est-il sûr ?
Le traitement (candidat médicament, dispositif médical…) est testé chez un petit groupe de personnes (le plus souvent des volontaires sains), pour la première fois, dans le but d'étudier comment il se comporte dans l'organisme humain.
L’essai de phase I a pour but de répondre aux questions suivantes :
- Quelle est la dose maximale tolérée (études de tolérance et de toxicité) ?
- Comment est-il assimilé dans l'organisme : administration, diffusion, métabolisme (étude de biodisponibilité ou de pharmacocinétique clinique) ?
- Quels en sont les effets secondaires dans ces conditions d'utilisation ?
Essai de phase II : A quelle dose le produit a-t-il un effet bénéfique avec une bonne tolérance ?
L’essai de phase II est mené sur un petit groupe homogène de volontaires atteints de la maladie ciblée.
L’objectif est de rechercher la plus petite dose avec la meilleure efficacité et la meilleure tolérance pour déterminer la dose optimale pour l’essai de phase suivante (phase III).
Essai de phase III : le produit a-t-il un effet thérapeutique sur un plus grand nombre de participants ?
L’essai de phase III se fait sur une plus grande population de volontaires atteints de la maladie ciblée.
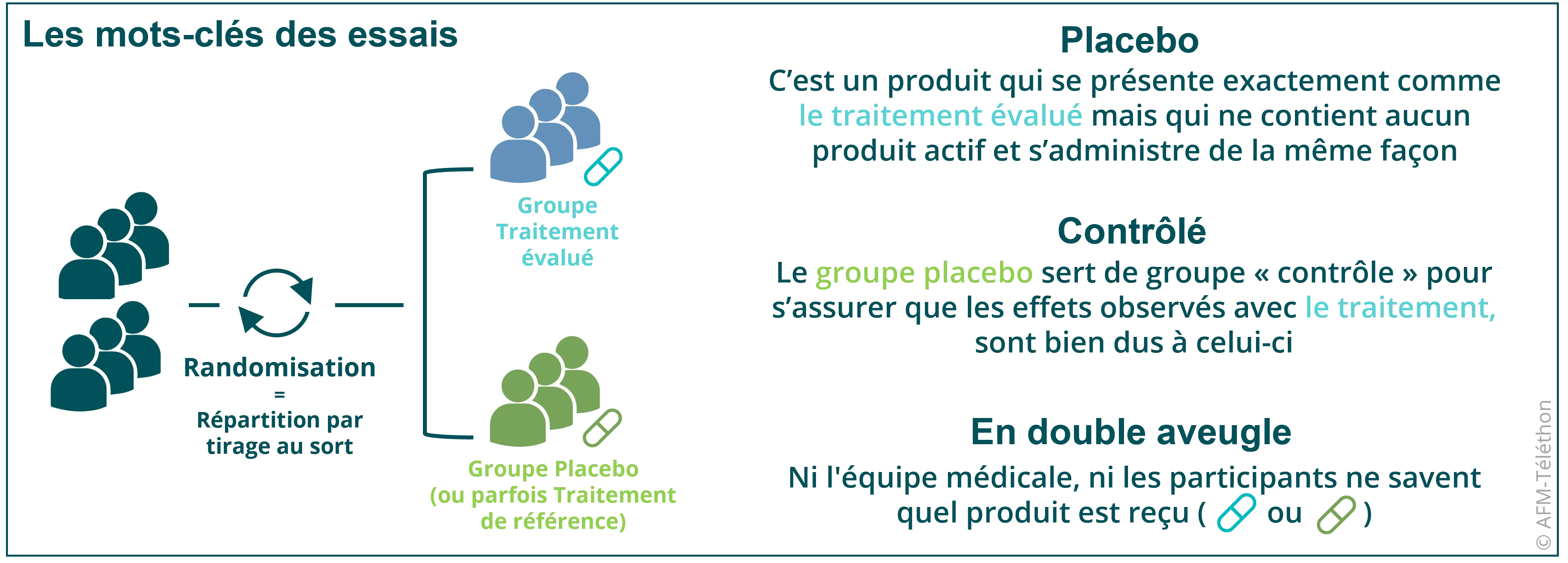
Le but est de confirmer la tolérance et l’efficacité du candidat médicament (en comparant son efficacité à celle d’un placebo ou d’un traitement existant).
Il permet également de déterminer dans quelles conditions d'utilisation le produit est le plus efficace.
Ce n’est que si l'analyse statistique des données recueillies a pu démontrer une efficacité suffisamment importante du candidat médicament, qu’un dossier de demande d'Autorisation de Mise sur le Marché (AMM) est constitué auprès de l’agence nationale de sécurité du médicament et des produits de santé (ANSM).
A noter que dans le cas des maladies rares et de thérapies innovantes (comme la thérapie génique), plusieurs phases peuvent coexister dans un même essai. On pourra parler d’essai de phase I/II puis de phase II/III par exemple. Le principe reste le même : les premiers essais ayant pour objectif principal de s’assurer de la bonne tolérance et les suivants de confirmer l’efficacité du traitement.
Essai de phase IV : phase de pharmacovigilance
L’essai de phase IV est réalisé après la mise sur le marché du médicament. Cette phase de pharmacovigilance se déroule dans les conditions habituelles d'emploi définies par l'Autorisation de Mise sur le Marché du médicament.
L’objectif d’un essai de phase IV est d'affiner la connaissance du médicament dans la pratique clinique (risques, bénéfices, conditions d'utilisation optimales), de mieux évaluer sa place dans la stratégie thérapeutique de la maladie ciblée et d'argumenter la révision de l'AMM.
Tant que le médicament est disponible sur le marché, les études de phase IV ne sont jamais terminées.
Participer à un essai, un choix personnel
Participer à un essai clinique est un choix très personnel. Il doit se faire après mûre réflexion, en ayant reçu toutes les informations et posé toutes les questions nécessaires.
Avant de prendre une décision, une note d'information écrite reprenant les informations sur le déroulement de l’essai est remise.
N’hésitez pas à interroger le médecin investigateur pour obtenir toutes les informations nécessaires, reformulez avec lui ce que vous avez compris, parlez-en à d'autres personnes (un autre médecin, votre famille, un psychologue…).
Quelques questions à ne pas oublier :
- Quel est l’objectif de l'essai ?
- Pourquoi pense-t-on que le produit testé peut être efficace ?
- Quels bénéfices sur la maladie peut-on en attendre ?
- Comment se déroulera l'essai (visites, examens médicaux…) ? Est-il contraignant ?
- Quels sont les risques éventuels, les effets secondaires possibles ?
- Quel sera le suivi médical pendant l'essai ? Et après l'essai ?
- Existe-t-il une assurance ?
- Comment pourrai-je m'organiser les jours de visite ? Qui pourra m'aider à mettre en place cette organisation ?
- Les frais seront-ils remboursés et par qui ?...
Il est important de bien estimer les contraintes, notamment logistiques (fréquence des visites, longueur des trajets...), que peut entrainer la participation à un essai.
Devenir le participant d’un essai clinique c’est entrer dans un protocole rigoureusement défini tant pour assurer la sécurité des participants, que pour obtenir un résultat fiable et pertinent. C’est aussi accepter des contraintes de visites et d’examens à l’hôpital supplémentaires.
L'entrée dans un essai clinique n'est possible qu’à partir du moment où l’essai a obtenu :
- l’autorisation de l'Agence nationale de sécurité du médicament et des produits de santé (ANSM), garante de la sécurité sanitaire de tout essai clinique en France ;
- l'avis favorable du Comité de protection des personnes (CPP), composé de professionnels de la santé et de représentants de la société civile (dont les associations de malades) et qui se prononce sur l'intérêt et les risques de l’essai clinique.
Le consentement éclairé
Aucune participation à un essai clinique n’est obligatoire. Toute personne est entièrement libre d’accepter ou de refuser de participer à un essai, sans avoir à se justifier et sans que cela nuise à son suivi médical ou à sa relation médecin/patient.
L’accord de participation à l’essai est formalisé par la signature d’un formulaire de consentement. En signant ce document, la personne atteste aussi qu’elle a bien reçu les informations nécessaires avant de prendre sa décision.
Même après avoir donné son consentement par écrit, il est possible à tout moment de se retirer de l’essai.
Des critères d’inclusion et de non inclusion
Les participants à une étude clinique doivent répondre à des critères très précis (d’âge, de sexe, de diagnostic, d’antécédents médicaux...), définis dans le protocole et propres à chaque étude.
Pour décider de l’inclusion d’une personne dans l’essai, le médecin investigateur, responsable de la conduite de l’essai dans un centre où se déroule l’essai (centre investigateur), se base sur les critères définis dans le protocole et s’assure que la personne y répond bien.
La visite dite « d’inclusion » au centre investigateur permet de vérifier les critères dont la plupart sont évalués par des examens médicaux (capacité vitale, mesure de la force musculaire...).
Les critères d’inclusion : qualité de l’essai et sécurité des patients
- Définis dans le protocole de chaque essai, les critères d’inclusion caractérisent la population de malades qui pourra y participer. Ils sont stricts et permettent de former des cohortes de participants relativement homogènes, ce qui est indispensable pour interpréter les résultats de l’essai. Ils sont décidés en fonction du produit, de la maladie, de son évolution… et de ce que la recherche veut montrer.
- Ce sont par exemple l’anomalie génétique en cause, l’âge, le sexe, des indicateurs fonctionnels moteurs (ambulant, non ambulant, distance de marche, …) et physiologiques (capacité respiratoire, état du cœur…).
Une fois inclus dans l’essai, le participant suit le traitement et se rend aux visites de suivi selon le planning établi pour cet essai clinique. La durée de cette période de participation à l'essai dépend de chaque essai.
Une fois la période de participation à l’essai terminée
À la fin d'un essai, le suivi médical lié à cet essai se termine et le malade reprend la prise en charge médicale qu’il avait avant d’entrer dans l’essai.
Dans certains cas, un protocole spécifique permet aux participants de prendre le candidat médicament en attendant que les autres participants aient terminé l’essai et que les résultats soient analysés et connus.
- En cas de résultats positifs, un accès précoce ou un accès compassionnel peuvent être délivrés pour pouvoir bénéficier du traitement en attendant l’obtention de l’autorisation de mise sur le marché (AMM).
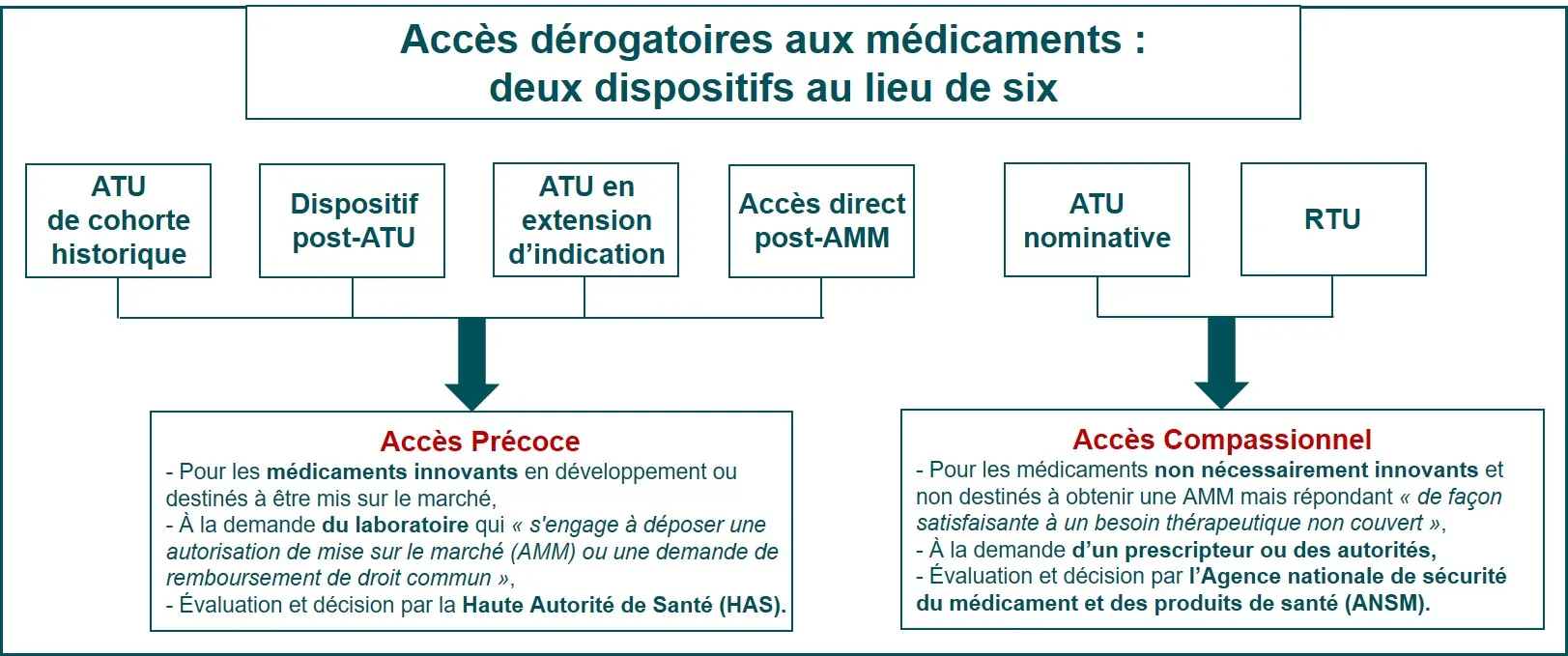
- En cas de résultats négatifs, le traitement est arrêté.
Parfois lorsque l’essai est terminé, on peut se sentir livré à soi-même, voire "abandonné".
Cette période de retour à une modalité de prise en charge antérieure peut être déstabilisante et nécessiter un accompagnement par l'équipe médicale, le Service régional de l’AFM-Téléthon, voire un psychologue.