Myosites (myopathies inflammatoires)
Découvrez les différentes formes de myosites, leurs symptômes, les progrès des traitements et les avancées de la recherche pour ces maladies auto-immunes musculaires.
Une myosite, c’est quoi ?
En médecine le suffixe « ite » désigne une inflammation, comme dans les mots otite ou bronchite. Une myosite est une inflammation de muscles (« myo »). Elle est dite idiopathique lorsque l’on ne retrouve aucune cause à cette inflammation : elle n’est pas dûe à une infection par un microbe comme les myosites virales secondaires par exemple à une grippe, ni à un problème de thyroïde... On parle également de myopathie, c’est-à-dire de maladie (« pathie ») du muscle, inflammatoire. L’inflammation est une réaction de l’organisme, via le système immunitaire, pour se protéger contre une agression, mais dans les myosites elle se produit en raison d'un dérèglement du système immunitaire.
En pratique une myosite associe la plupart du temps :
- les signes d’une atteinte musculaire : faiblesse, douleur musculaire, augmentation de la créatine phosphokinase (CPK ou CK) dans le sang
- des signes que cette myopathie est d’origine immunitaire : inflammation des fibres musculaires, production d’anticorps dirigés contre des constituants du corps (ou auto-anticorps)
Des maladies musculaires rares
L'ensemble des myopathies inflammatoires ou myosites toucheraient 1 personne sur 7 000. En France, au moins 7 000 adultes et enfants seraient touchés par une forme de myosite. Il s’agit plus souvent de filles ou de femmes que de garçons et d’hommes, sauf pour la myosite à inclusions qui touche deux fois plus d’hommes que de femmes.
Un dérèglement du système immunitaire
Le rôle du système immunitaire est de protéger l'organisme contre des agressions extérieures, comme les infections. Dans une myopathie inflammatoire, il se dérègle et produit des auto-anticorps c’est-à-dire des anticorps qui attaquent certains constituants de l'organisme, comme les muscles. Une myosite est une maladie auto-immune, à l’instar de la polyarthrite rhumatoïde ou de la myasthénie.
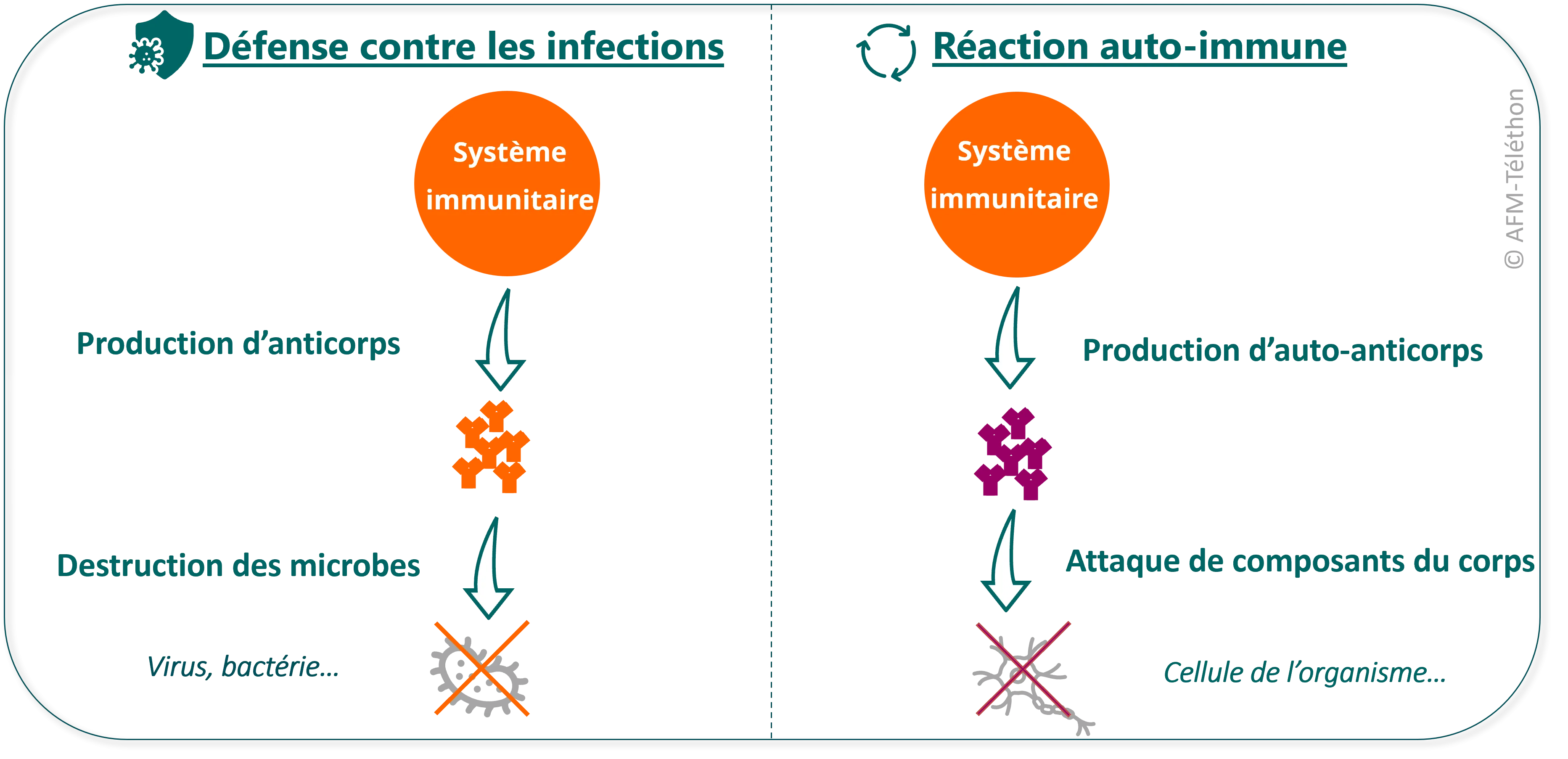
Mais pourquoi le système immunitaire se dérègle-t-il dans les myosites ? Médecins et chercheurs ne le savent pas encore précisément. Leur hypothèse actuelle est que la maladie résulte probablement de la conjonction de facteurs liés à l’environnement (rayons ultraviolets, tabac, infections virales, prise d’un médicament...) et d’un terrain génétique prédisposant. Néanmoins les myosites ne sont pas des maladies héréditaires, qui seraient liées à une anomalie génétique. Elles ne sont donc pas transmissibles à ses enfants.
Les différents types de myosites
Les symptômes, le type d’auto-anticorps (immunologie) et l'aspect du tissu musculaire au microscope (histologie) permettent de distinguer aujourd’hui cinq formes principales de myopathies inflammatoires.
La myosite à inclusions
- La myosite à inclusions dite « sporadique (car non génétique) ne touche que les adultes. C’est la plus fréquente des myopathies passé l’âge de 50 ans.
- Elle débute toujours après 30 ans, le plus souvent après 50 ans, par une faiblesse des muscles des cuisses (quadriceps) et des doigts (fléchisseurs) qui évolue de façon lente et progressive. Les muscles qui servent à déglutir ainsi que ceux qui aident à maintenir droit le dos et le cou sont parfois touchés également.
- La maladie associe une inflammation et un vieillissement prématuré (dégénérescence) des cellules musculaires. L’examen au microscope d’un prélèvement de muscle (biopsie) montre ces deux phénomènes, avec présence d’agrégats de protéines toxiques (les « inclusions »).
- Le diagnostic de myosite à inclusions peut être long et compliqué. Un groupe international d’experts a récemment revu et corrigé la liste des critères nécessaires pour établir ce diagnostic, avec l’objectif de le faciliter et de l’accélérer.
- Les médicaments habituels des myosites (corticoïdes, immunosuppresseurs) sont inefficaces dans la myosite à inclusions, mais de nouveaux traitements sont à l’essai.
Voir les Avancées dans les myopathies inflammatoires
La dermatomyosite
- Dans la dermatomyosite, le système immunitaire s’attaque aux petits vaisseaux sanguins qui irriguent les muscles (faiblesse des épaules, des cuisses…) et la peau (rougeur ou éruptions du visage, des mains, du haut du dos…).
- La maladie peut débuter dans l’enfance (dermatomyosite juvénile, en moyenne autour de l’âge de 7 ans) ou à l’âge adulte. C’est la plus fréquente des myosites, tous âges confondus.
- Elle s’associe à un risque plus élevée de développer un cancer (20 à 30% des patients) dans les trois ans qui précèdent ou qui suivent le diagnostic. Ce risque concerne quasi exclusivement les adultes, chez lesquels un bilan régulier est réalisé pour dépister une tumeur débutante éventuelle, dont les chances de guérison sont alors maximales.
La myopathie nécrosante auto-immune
- Identifiée en 2003, la myopathie nécrosante auto-immune peut débuter chez l’enfant comme chez l’adulte mais commence souvent vers l’âge de 40 ans. Elle touche uniquement les muscles, se manifestant par des douleurs et une faiblesse musculaire (épaules, hanches…) progressive.
- Elle est parfois provoquée par un médicament pris pour faire baisser le taux de cholestérol appartenant à la famille des statines. Les symptômes apparaissent alors souvent deux à trois ans après le début du traitement, mais persistent ou s’aggravent après son arrêt.
- L’examen au microscope de la biopsie musculaire montre une nécrose (destruction) importante des fibres mais, contrairement aux autres myosites, peu ou pas d’inflammation, ce qui peut compliquer le diagnostic.
La polymyosite
- Débutant toujours après l’âge de 18 ans, souvent entre 20 et 50 ans, la polymyosite se manifeste par une atteinte isolée des muscles, souvent ceux de la racine des membres (épaules, hanches).
- Dans cette maladie, des cellules immunitaires (les lymphocytes T cytotoxiques ou CD8+) attaquent les fibres musculaires.
- Grâce au progrès des connaissances et à la découverte de nouveaux types de myosites, on sait que la polymyosite est désormais la moins fréquente des myopathies inflammatoires de l’adulte. Les personnes chez qui ce diagnostic a été posé par le passé auraient en fait souvent une autre forme de myosite, par exemple un syndrome des antisynthétases ou une myopathie nécrosante auto-immune. Une étude dont les résultats sont parus en 2024 l’a confirmé.
Les myosites de chevauchement
- Une myosite de chevauchement peut commencer de façon insidieuse ou évoluer rapidement, dans l’enfance comme à l’âge adulte.
- Le mot « chevauchement » exprime le fait que la myosite associe une atteinte des muscles ou de la peau à des signes ou à des auto-anticorps présents dans une autre maladie (lupus, polyarthrite, pneumopathie interstitielle…). Les manifestations peuvent donc être très différentes d’une personne à l’autre : douleurs et faiblesse musculaires, peau des doigts épaisses et fissurées, troubles de la circulation des extrémités (syndrome de Raynaud), difficultés à avaler, à respirer, douleurs des articulations…
- Le syndrome des antisynthétases fait partie des myosites de chevauchement. Il touche les muscles, les poumons, la peau et les articulations, avec présence d’auto-anticorps dirigés contre les aminoacyl-ARNt synthétases (anti-Jo1, anti-PL7, anti-PL12…) d’où son nom.
- De même, les médecins ont identifié récemment la scléromyosite. Elle comporte des caractéristiques de myosite et des éléments de sclérodermie, une autre maladie auto-immune.
La classification se précise au fil du temps
Les progrès de la science et des connaissances font évoluer la classification des myosites d’année en année.
• La « dermato-polymyosite » a longtemps été le seul type de myopathie inflammatoire reconnu.
• En 1975, les médecins distinguaient la dermatomyosite et la polymyosite.
• Ils différencient cinq types de myosites depuis 2005 et la reconnaissance des myosites de chevauchement.
• Ils pourraient à l’avenir n’en retenir que 4, si l’existence en tant que tel de la polymyosite venait à être remise en cause.
Quels examens pour faire le diagnostic ?
Le diagnostic de myosite s’appuie sur un ensemble d’arguments : l’âge de début, les symptômes ressentis, leur évolution, les signes retrouvés à l’examen par le médecin et les résultats de différents examens complémentaires, le fait qu’aucun autre membre de la famille ne présente ou n’a présenté les mêmes symptômes...
- Une prise de sang permet de doser la créatine phosphokinase (CPK ou CK pour créatine kinase), une enzyme musculaire dont le taux augmente souvent en cas de myosites.
- L’électromyogramme éventuel fait évoquer une myopathie inflammatoire, mais seule l’examen au microscope de la biopsie musculaire peut permettre au médecin de l’affirmer avec certitude. Il est parfois nécessaire de la répéter.
- Le médecin peut demander une imagerie par résonance magnétique, ou IRM, pour l’aider à choisir le site de la biopsie musculaire dans une zone où la maladie est repérée comme active sur les images : augmentation de volume du muscle traduisant un œdème, zones apparaissant plus « blanches » en raison d’une inflammation.
- La recherche de différents auto-anticorps dans le sang conforte le diagnostic et l’affine, permettant souvent de savoir quel est le type de myosite en cause.
Un bilan sur-mesure
Les examens demandés sont variables selon le patient et ses symptômes. Par exemple, la biopsie musculaire n’est pas toujours pratiquée lorsqu’existe sur la peau des signes typiques de dermatomyosite.
Les enzymes musculaires, quel rôle diagnostique ?
Les CPK (créatine phosphokinase) ou CK (pour créatine kinase) sont des enzymes normalement présentes à l’intérieur des cellules musculaires. Leur taux augmente dans le sang lorsque ces cellules sont abimées ou détruites, par exemple en cas de maladie du muscle, mais aussi après une activité physique intense, une biopsie musculaire ou un électromyogramme.
Dans les différentes myosites, le taux de CPK dans le sang peut augmenter. Par exemple, il atteint parfois dix fois la normale au début d’une myopathie nécrosante auto-immune. De même, le taux de CPK est souvent augmenté dans la myosite à inclusions, de façon modérée : il reste inférieur ou égal à 15 fois la normale, un seuil qui fait désormais partie des critères diagnostiques de cette maladie.
Toute augmentation du taux de CPK oriente fortement le diagnostic vers une myopathie, sans pour autant savoir de quelle type de maladie musculaire (inflammatoire, génétique…) il s’agit. C’est un élément du diagnostic mais il ne suffit pas à lui seul. Son dosage peut être répété, pour éliminer une cause d’augmentation transitoire.
Le taux de CPK peut…
• être normal dans une myosite, y compris en phase active,
• se normaliser au cours de l’évolution de la maladie soit parce qu’elle n’est plus en phase activen grâce au traitement, sans en raison d’une perte importante de masse musculaire.
Le médecin peut demander le dosage d’autres enzymes musculaires, comme la lactate-déshydrogénase (LDH) et l'aldolase.
Les différents auto-anticorps dans les myosites
Deux grandes familles d’auto-anticorps peuvent être produits par l’organisme en cas de myosite :
- les auto-anticorps spécifiques des myosites sont retrouvés uniquement chez les personnes atteintes de ces maladies (antiJo-1, anti-Mi2, anti-SRP, anti- HMGCR, anti-TIF1-γ…),
- les auto-anticorps associés aux myosites peuvent être présents aussi dans d’autres maladies auto-immunes comme le lupus (antiRo-52, anti-Ro60, anti-La, anti-PM-Scl 75…).
Certains auto-anticorps orientent vers tel ou tel type de myosite. Ainsi, les auto-anticorps anti-SRP et anti-HMGCR signent l’existence d’une myopathie nécrosante auto-immune. Les anti-SAE, anti-TIF1-γ, anti-NXP-2 et anti-Mi2 et anti-MDA-5 sont considérés comme spécifiques de la dermatomyosite. Pour les anti-cN1A, c’est plutôt une myosite à inclusions. À l’inverse, on ne retrouve aucun auto-anticorps spécifique ou associé aux myosites dans la polymyosite.
Parfois pas d’auto-anticorps détecté
La recherche d’auto-anticorps peut revenir « négative » : ce serait le cas pour près de 40% des personnes qui ont pourtant bien une myosite. Cela ne signifie pas forcément qu’elles ne produisent pas d’auto-anticorps. Elles peuvent par exemple produire un type d'auto-anticorps non encore identifié à ce jour. Une myosite auto-immune pourrait se déclencher également sans qu'aucun auto-anticorps ne soit produit.
Quelle est l’évolution d’une myosite ?
En l’absence de traitement, une myosite ne s’améliore pas de façon spontanée. La faiblesse musculaire s’aggrave progressivement et peut s’étendre à d’autres muscles, avec le risque notamment d’une perte de la marche, mais aussi des risques possiblement vitaux pour certaines atteintes : difficultés à avaler et son risque de fausses routes et d’infections pulmonaires, insuffisance respiratoire, atteinte du muscle cardiaque…
Sous traitement, la plupart des myosites s’améliorent avec l’obtention d’une rémission. Certaines personnes connaissent une récupération, complète ou incomplète, sans récidive ultérieure et dans certains cas pourront arrêter tout traitement. D’autres connaitront une nouvelle poussée de la maladie (rechute) pendant les premiers mois ou années qui suivent, mais ce risque concerne une minorité de patients.
Pour réduire le risque de rechute, le traitement initial est habituellement suivi d’un traitement dit « d’entretien » pendant au moins deux à trois ans (en moyenne), au cours desquels les doses de médicaments sont peu à peu réduites.
La myosite à inclusions occupe une place à part puisqu’elle est peu ou pas sensible au traitement habituel des myosites. Néanmoins, elle évolue de façon très lente, sur des années, et ne modifie pas l’espérance de vie qui reste comparable à celle d’une personne indemne de myosite.
En savoir plus sur les myosites avec une Web conférence des associations de patients
Le traitement de fond des myosites
La prise en charge d’une personne atteinte de myopathie inflammatoire associe souvent un traitement de fond à des mesures pour agir sur la force musculaire et prévenir les complications. Hormis pour la myosite à inclusions, le traitement de fond repose sur des médicaments qui diminuent ou modulent l’activité du système immunitaire et l’inflammation, avec l’objectif de modifier l’évolution de la maladie.
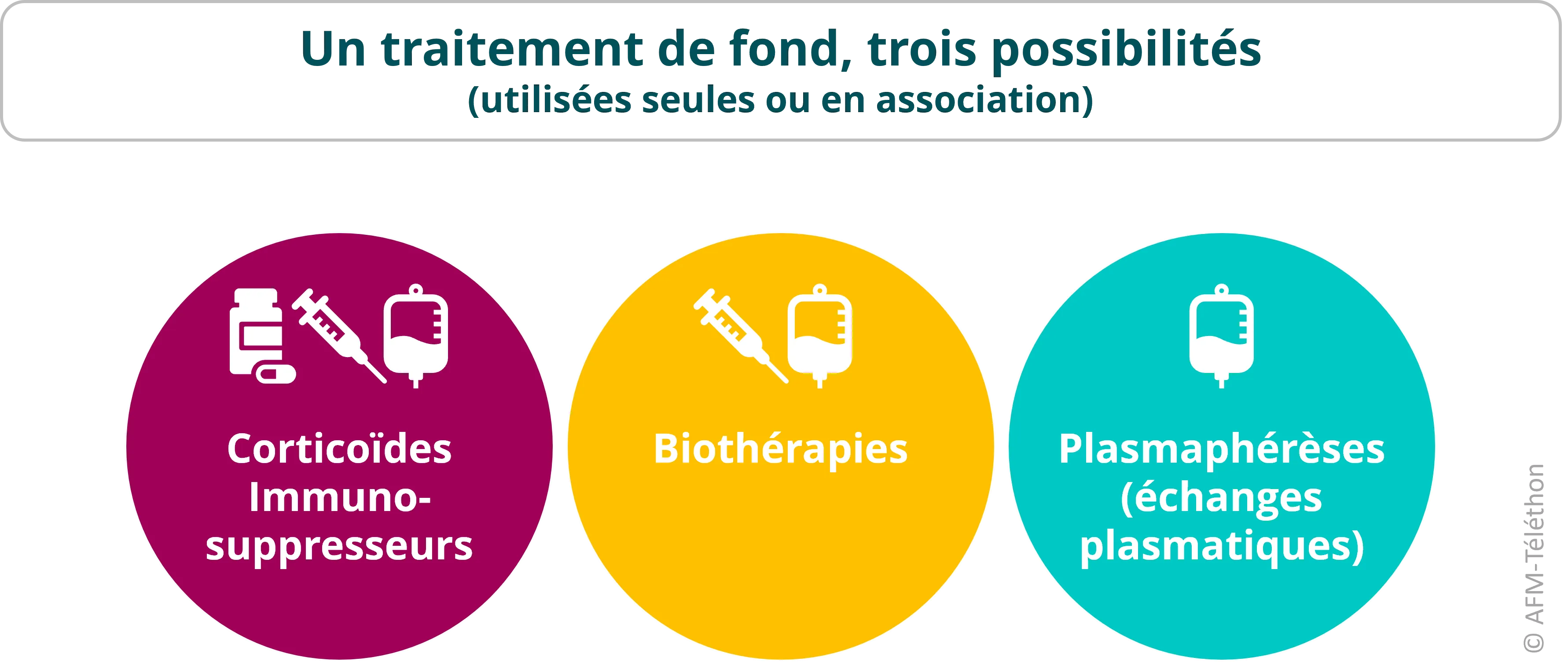
- Les corticoïdes comme la prednisone réduisent l’inflammation du muscle (effet anti-inflammatoire) et, à forte dose, diminuent la réponse immunitaire (immunosuppresseur).
- Les immunosuppresseurs comme le méthotrexate, l’azathioprine, le cyclophosphamide, le tacrolimus ou encore la rapamycine (sirolimus) diminuent fortement l’activité du système immunitaire, raison pour laquelle ils sont également prescrits après une greffe d’organe pour réduire le risque de rejet.
- Les biothérapies sont produites à partir d'une source biologique (cellules, bactéries…). Les immunoglobulines dites « polyvalentes » en font partie : ce sont des anticorps produits par les cellules immunitaires de donneurs en bonne santé. Le plus souvent administrées par voie intraveineuse, elles sont capables de moduler l’activité du système immunitaire (immunomodulateur) dans les myosites.
Différents anticorps monoclonaux (rituximab, inhibiteurs des janus kinases…) sont également des biothérapies. On parle dans ce cas également de « thérapies ciblées » car chacun de ces anticorps est dirigé contre une cible biologique spécifique, par exemple une protéine impliquée dans l’inflammation. Ils exercent une action immunosuppressive ou immunomodulatrice sélective.
- La plasmaphérèse (ou échange plasmatique) consiste à filtrer, à l’aide d’une machine, le sang afin d’en éliminer les auto-anticorps.
Le médecin choisit le ou les moyens de traitement les plus adaptés, au cas par cas, en fonction du type de myosite, des atteintes associées (poumons, peau...) et des autres problèmes de santé qui peuvent faire contre-indiquer certains médicaments.
Le traitement de fond de l’inflammation des muscles contribue aussi à améliorer la plupart des atteintes d’autres organes (peau, cœur, poumons…), qu'elles soient liées à la myosite elle-même ou à une autre maladie auto-immune associée. Un traitement complémentaire est parfois nécessaire.
Le saviez-vous ?
En agissant sur le système immunitaire, les corticoïdes, immunosuppresseurs et thérapies ciblées diminuent également ses capacités de défense contre les infections. Le risque de contracter une infection est augmentée pour les personnes qui prennent ce type de traitement et cette infection risque de surcroit de provoquer une poussée de myosite ou d’en augmenter l’intensité. C’est pourquoi la vaccination contre la grippe notamment, chaque année à l’automne, est fortement recommandée avec ces médicaments.
Quelles options pour les formes résistantes aux médicaments habituels ?
Dans la majorité des cas, le traitement de fond débute par des corticoïdes, seuls ou associés dès le départ à un médicament immunosuppresseur, à la maison ou à l’hôpital, en fonction de l’intensité des manifestations de la maladie.
Pour les patients dont les symptômes ne s’améliorent pas ou pas assez avec ce premier traitement (myosite dite « réfractaire »), d’autres médicaments sont prescrits : association de deux immunosuppresseurs et/ou immunoglobulines polyvalentes et/ou thérapie ciblée.
Un grand nombre de médicaments, et notamment de thérapies ciblées, sont en développement ou à l’essai dans les myosites réfractaires aux traitements usuels.
Voir les essais et études cliniques en France dans les myosites
Prise en charge multidisciplinaire et suivi médical
La prise en charge et le suivi d’une myosite se conçoit au mieux dans le cadre de consultations pluridisciplinaires spécialisées dans les maladies neuromusculaires ou les maladies auto-immunes rares. Elles réunissent, dans une même structure hospitalière, les compétences de plusieurs intervenants (interniste, neurologue, médecin de rééducation, kinésithérapeute, diététicien, psychologue…) et sont structurées selon deux dispositifs :
- la filière de santé des maladies rares neuromusculaires Filnemus,
- la filière de santé des maladies auto-immunes et auto-inflammatoires rares FAI2R.
Un centre expert national
En décembre 2023, l'Hôpital de la Pitié-Salpêtrière (équipe du Pr Olivier Benveniste) est devenu le Centre de référence des myopathies inflammatoires de la filière Filnemus. C'est le seul en France à être dédié aux myosites. Sa labellisation est une reconnaissance de l'expertise et de l'importance de l'activité du centre parisien sur le plan tant du diagnostic et de la prise en charge que de la recherche (essais cliniques notamment).
Le suivi médical doit débuter dès le diagnostic de myosite évoqué. La fréquence des consultations et des bilans est adaptée à chaque personne, en fonction du type de myosite, du traitement instauré, des atteintes associées éventuelles et de l’évolution de la maladie.
Les soins courants et les urgences
Quel que soit le type de myopathie inflammatoire dont vous êtes atteint, il est essentiel d’en informer tous les professionnels de santé consultés (médecins, dentistes, chirurgiens, pharmaciens…) et de leur dire quels sont vos traitements en cours. La prise d’un immunosuppresseur, de corticoïdes ou d’une biothérapie nécessite en effet de prendre des précautions particulières.
N’hésitez pas à parler à vos professionnels de santé non experts en maladies neuromusculaires, de différents documents qui peuvent les aider à s’informer :
- le Protocole national de diagnostic et de soins (PNDS) de la dermatomyosite de l’enfant et de l’adulte, qui explique la prise en charge optimale dans cette maladie et s’accompagne d’une synthèse à destination du médecin traitant,
- le PNDS de la myosite à inclusions sporadique et sa synthèse destinée au médecin traitant,
- la fiche « Bonnes pratiques en cas d’urgence » dans la dermatomyosite, accessible sur le site Orphanet,
- la Carte d’urgence Dermatomyosite, distribués par les médecins.
S’informer, pour mieux se soigner
Élaboré par l’AFM-Téléthon avec le concours d’experts et de patients, le Zoom sur… les myopathies inflammatoires présente une information détaillée sur ces maladies. Comment se manifestent-elles et évoluent-elles ? Quels sont les traitements possibles et leurs effets indésirables potentiels ? À quelle fréquence consulter ? Quid du désir d’enfant avec une myosite ? Quelles démarches administratives ? ... À toutes ces questions, il apporte des réponses précises et concrètes, utiles au quotidien.
Kinésithérapie et activité physique
L’exercice physique est une composante essentielle du traitement de toutes les formes de myosites, avec l’objectif d’améliorer la force musculaire.
Un programme personnalisé de rééducation motrice, en kinésithérapie, permet de faire travailler tous les muscles. En complément, il est recommandé de pratiquer une activité physique ou sportive adaptée.
Les séances de kinésithérapie contribuent également à lutter contre les douleurs et à prévenir les rétractions musculo-tendineuses.
En savoir sur l’activité physique adaptée (avec démonstration)
Recherche et progrès scientifiques
Chaque année, des centaines de publications médicales et scientifiques témoignent de l’intensité des travaux de recherche sur les myosites, qui mobilisent dans le monde de nombreuses équipes de chercheurs et de médecins. En France, l'équipe de recherche Myopathies inflammatoires & thérapies innovantes ciblées de l’Institut de myologie (Hôpital de la pitié Salpêtrière, Paris) est très active en ce domaine.
En chiffres
- 120 essais et études cliniques en cours ou en préparation dans les myosites sont répertoriés sur le site ClinicalTrials.gov dont 22 en France (interrogation du 19 juin 2025).
- Plus de 1 600 articles sur les myosites publiés dans la littérature médicale et scientifique en un an (interrogation de Pubmed le 19 juin 2025)
Les résultats obtenus par les différentes équipes de recherche contribuent de façon très concrète à améliorer le diagnostic des myosites et leur traitement.
Voir les Avancées dans les myopathies inflammatoires
Des mécanismes de mieux en mieux connus
De nombreux travaux de recherche s’attachent à améliorer la compréhension des mécanismes des myosites, ce qui peut ouvrir de nouvelles pistes de traitement. Ainsi, la découverte, ces dernières années, que la dermatomyosite était une maladie des interférons a abouti à évaluer dans cette maladie l’efficacité d’inhibiteurs des janus kinases, des médicaments qui ciblent les interférons.
Parmi les dernières avancées en ce domaine également :
- certaines études ont identifié des facteurs génétiques ou environnementaux qui favorisent l'apparition d'une myosite.
- d’autres ont prouvé le rôle pathologique des auto-anticorps (anti-SRP et anti-HMGCR dans la myopathie nécrosante auto-immune), évalué leur intérêt pour le diagnostic ou dans le cadre du suivi.
- d’autres enfin ont précisé les symptômes possibles.
De nouveaux traitements à l’essai
Les traitements immunosuppresseurs habituellement utilisés dans les myopathies inflammatoires ne sont pas toujours efficaces (myosite à inclusions, myosites réfractaires). Il est donc nécessaire de développer d'autres approches.
Les cellules CAR-T combinent thérapie cellulaire et thérapie génique. Mi-2025, elles faisaient l'objet de 29 essais cliniques dans le monde, pour différents types de myopathies inflammatoires. Deux de ces essais ont lieu en France.
La thérapie cellulaire commence également à être explorée, avec notamment deux essais en cours dans la myosite à inclusions, dont l'un en France, soutenu par l'AFM-Téléthon.
Différents médicaments sont également en cours d'évaluation :
- des produits déjà commercialisés pour soigner d’autres maladies, comme les inhibiteurs des janus kinases (baricitinib, tofacitinib, ruxolitinib...), la rapamycine, les anticorps anti récepteurs Fc néonataux (efgartigimod…) ou encore le thiosulfate de sodium.
- des molécules encore en développement, comme l'ulviprubart (ABC008) pour la myosite à inclusions et le GLPG3667 dans la dermatomyosite, tous deux à l'essai en France.
Voir les essais et études cliniques en France dans les myosites
Des soutiens utiles
Les myosites font partie des maladies neuromusculaires dont l’AFM-Téléthon suit et finance des travaux de recherche. L’association accompagne aussi, concrètement, les malades et leurs familles via :
- ses Services régionaux et leurs Référents parcours de santé (RPS) qui vous aident à trouver et mettre en œuvre des réponses à vos préoccupations (suivi médical, aide humaine, intégration scolaire, emploi...) et vous accompagnent dans vos démarches,
- son Groupe d’Intérêt Myopathies Inflammatoires (GIMI) qui diffuse des informations sur les myosites et rompt l’isolement des personnes qui en sont atteintes au travers de témoignages, de conseils et d’un soutien moral,
- ses Délégations départementales qui rassemblent des bénévoles concernés par une maladie neuromusculaire et assurent une proximité et un lien auprès des familles.
Vous souhaitez en savoir plus ou avez une question particulière ? La ligne Accueil Familles de l’AFM-Téléthon est à votre écoute au 0 800 35 36 37 (appel gratuit).



Comment se manifestent-elles ?
Les myopathies inflammatoires (myosites) sont des maladies rares dont les manifestations possibles sont très diverses, selon leur type et selon les personnes. De ce fait, leur diagnostic prend parfois un certain retard.
Seules trois formes de myosites peuvent survenir chez un enfant ou un adolescent : la dermatomyosite, la myopathie nécrosante auto-immune et la myosite de chevauchement.
Des symptômes musculaires variables
Les myosites entrainent le plus souvent une faiblesse musculaire, une plus grande fatigabilité à l’effort et des douleurs des muscles (myalgies) ressenties comme des courbatures ou des crampes. Elles s’installent souvent de façon progressive, sur plusieurs semaines ou mois.
Les muscles touchés varient selon le type de myosite et la personne. L’inflammation peut concerner de préférence les muscles des hanches et des cuisses (comme le quadriceps), des bras et des épaules : ce sont les muscles dits « proximaux ». Dans d’autres cas, elle se localise plutôt au niveau des extrémités (muscles distaux) : mollets, pieds, doigts et poignets.
Il peut aussi exister une atteinte des muscles du visage, du dos, du cou, de la gorge (déglutition) ou encore des muscles respiratoires.
Ces différentes localisations ont des retentissements différents sur les activités du quotidien.
Les manifestations possibles
Selon les muscles touchés, la faiblesse musculaire provoquée par une myosite peut se traduire par des difficultés ou une gêne inhabituelle pour :
• se lever d’une chaise ou des toilettes sans l’aide des mains,
• monter les escaliers, gravir une échelle,
• se coiffer,
• se laver les cheveux,
• s’habiller,
• s’accroupir ou se relever de la position accroupie ou à genou,
• passer de la position couchée à la position assise,
• maintenir les mains au-dessus de la tête de façon prolongée,
• courir, marcher (enfant qui demande à être porté plus souvent, chutes répétées),
• soulever ou porter les objets lourds (sac de course, valise…),
• prendre en main et manipuler des objets (outils, fourchette...),
• écrire,
• maintenir le dos et la tête droite,
• relever la tête,
• avaler certains aliments,
• respirer.
Parfois d’autres organes atteints
La dermatomyosite et les myosites de chevauchement s’accompagnent d’autres signes, également liés à un mécanisme auto-immun et qui peuvent concerner :