Myopathies distales : quoi de neuf en 2022 ?

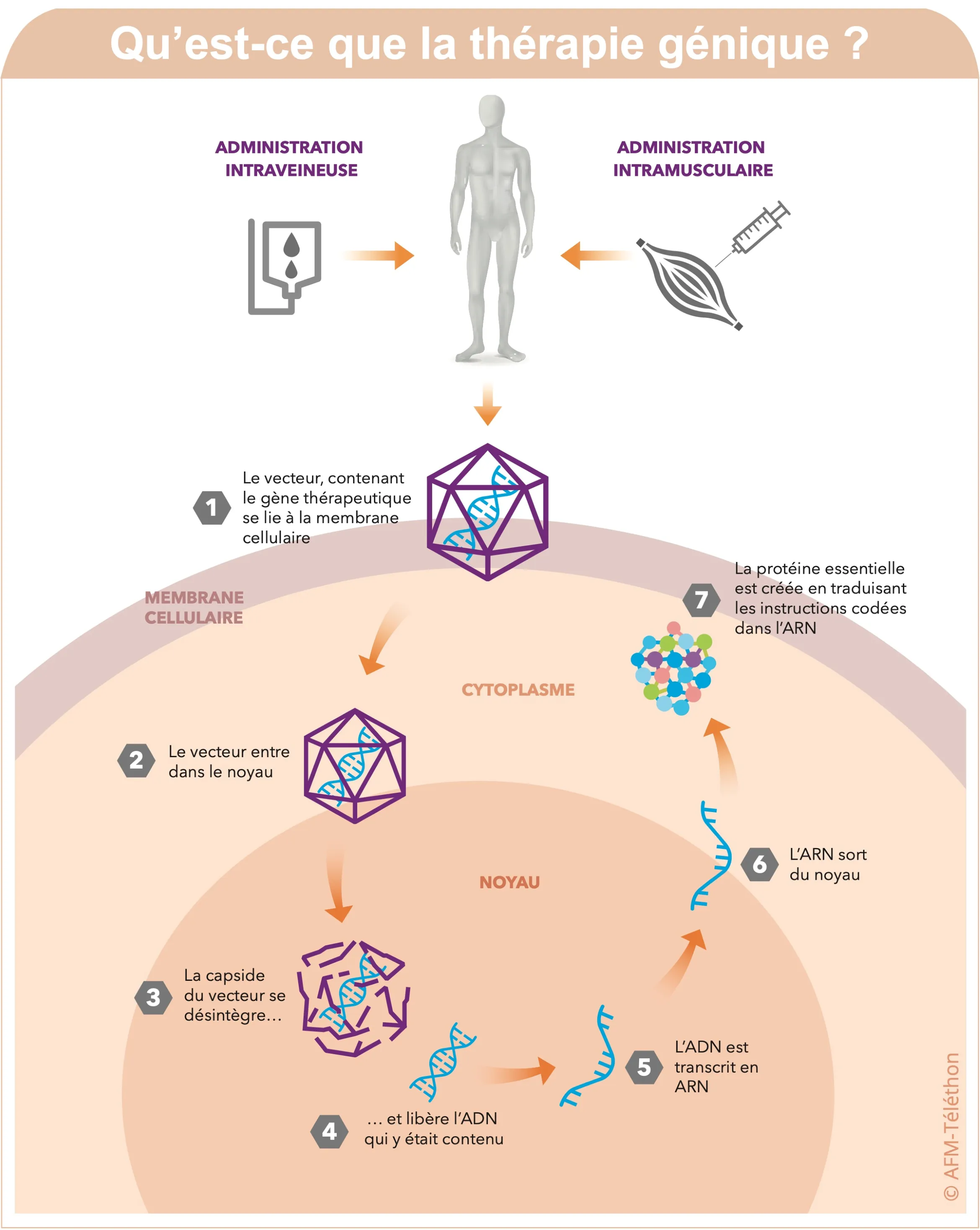
À l’essai clinique ou chez l’animal, les approches de thérapie génique se multiplient dans les myopathies distales. En parallèle, des approches pharmaceutiques plus classiques sont également à l’étude. D’autres études cliniques permettent d’améliorer le diagnostic et la prise en charge de ces maladies.
De la thérapie génique à la préparation des essais cliniques, une année riche d’enseignements pour la myopathie de Miyoshi
La myopathie de Miyoshi est liée à la dysferline. Un essai clinique en cours aux Etats-Unis vise à introduire, à l’aide d’un vecteur viral AAV, le gène de la dysferline dans le muscle de deux participants. Si les résultats de l’essai n’ont pas encore été publiés, l’injection intramusculaire chez les deux patients inclus n’a pas soulevé d’inquiétudes particulières quant à la sécurité d’utilisation du produit.

Deux autres approches de thérapie génique étudiées en laboratoire
La première consiste à apporter le gène de la sphingomyélinase acide lysosomale (ASM), une enzyme impliquée comme la dysferline dans la réparation de la membrane des cellules musculaires à l’aide d’un vecteur AAV.
Une injection unique du produit de thérapie génique à des souris atteintes de dysferlinopathie réduit la dégénérescence musculaire et permet de récupérer de la force musculaire.
Seconde approche : apporter le gène de la galectine-1 qui intervient à la fois dans la réparation membranaire et dans l’inflammation, deux mécanismes en jeu dans les dysferlinopathies. Les souris traitées ont une meilleure mobilité.
Cibler les anomalies génétiques de la dysferline
Constatant que les mutations de type non-sens sur le gène DYSF (qui code la dysferline) sont les plus fréquentes dans la population coréenne (plus de la moitié des malades présentent au moins une anomalie génétique de ce type), des chercheurs ont évalué l’intérêt thérapeutique de l’ataluren (ou Translarna), une molécule ciblant spécifiquement ces anomalies génétiques (translecture d’un codon stop).
Administré par voie orale pendant 2 semaines à des souris atteintes de dysferlinopathie avec mutations non-sens du gène DYSF, l’ataluren a réduit leur atteinte musculaire et amélioré leurs capacités motrices.
Un autre type de mutation sont les mutations faux-sens. Elles aboutissent à la formation d’une dysferline mal repliée et rapidement détruite. Elles représenteraient 30 à 40% des anomalies génétiques responsables d’une dysferlinopathie.
Le phénylbutyrate, un médicament utilisé dans le traitement de maladies du cycle de l’urée, améliore le repliement et/ou la stabilité des protéines. Apporté à des souris porteuses d’une anomalie faux-sens responsables d’une dysferlinopathie chez l’homme, le traitement rétablit l’activité de réparation membranaire de la dysferline dans les cellules musculaires.
Des études cliniques pour anticiper les essais cliniques à venir
Dans le cadre de l’étude internationale COS 2, des publications ont précisé les meilleurs critères d’évaluation en vue de futurs essais cliniques dans les dysferlinopathies. Plutôt que la mesure de la force musculaire, qui peut varier d’un jour à l’autre indépendamment de l’évolution de la maladie, les auteurs recommandent des IRM musculaires (imagerie par résonance magnétique) et notamment de séquences particulières (Séquence Dixon) permettant d'évaluer des modifications même minimes du remplacement graisseux des muscles des membres inférieurs.
L’étude Jain COS a permis d’établir des recommandations internationales pour le suivi respiratoire et cardiaque des personnes atteintes de dysferlinopathie. Si un examen respiratoire (mesure de la capacité vitale forcée) annuel ou biannuel est conseillé, les examens cardiaques ne sont utiles qu’en cas de signes d’une atteinte du cœur (palpitations, sensation de malaise…).
Un groupe de travail (ou « workshop ») organisé par l’European Neuromuscular Centre (ENMC) s’est réuni en juillet 2022 pour produire des recommandations sur la prise en charge des myopathies liées à la dysferline. Les docteurs Stojkovic, Evangelista et Fernández-Eulate de l’Institut de Myologie ont pu participer cette initiative et des recommandations seront rédigées prochainement.
Résultats prometteurs de thérapie génique dans d’autres myopathies distales
Dans la myopathie GNE (myopathie de Nonaka)
Outre les résultats encourageants de l’essai du ManNAc, montrant que le produit est bien toléré et semble efficace, une approche de thérapie génique a été étudiée chez des souris. L’injection d’un produit de thérapie contenant le gène GNE et ciblant les muscles a permis d’obtenir une fabrication durable de l’enzyme GNE dans les muscles des souris (jusqu’à un an et plus).
Dans la myopathie liée à TCAP
Un produit de thérapie génique (comprenant le gène TCAP dans un vecteur rAAV) a été testé chez des souris déficientes en TCAP entrainant une amélioration de l’atteinte musculaire, qui progresse plus lentement.
Des publications médicales nombreuses sur la prise en charge et le diagnostic des myopathies distales
Une nouvelle myopathie distale
Une équipe de l’Institut de Myologie (Paris) a montré, dans le cadre d’une collaboration internationale, l’implication du gène SMPX dans une myopathie distale liée à l’X chez 10 personnes atteintes d’une myopathie distale ayant débuté après l’âge de 30-40 ans.
L'implication du gène HNRNPA1 confirmée
L'équipe du Pr B. Udd, spécialiste international des myopathies distales, a mis en évidence une anomalie du gène HNRNPA1 chez 8 membres d'une famille atteints de myopathie distale débutant à l'âge adulte par une atteinte des muscles des membres supérieurs (au niveau des mains). C'est grâce à des techniques d'analyse du génome plus performantes que le diagnostic génétique a enfin pu être posé. Une précédente analyse génétique réalisée en 2004 n'avait pas permis de conclure sur le gène en cause.
Des troubles du sommeil et sanguins dans la myopathie GNE
Plusieurs articles médicaux présentent les atteintes non musculaires qui peuvent être présentes chez les personnes atteintes de myopathie GNE : des difficultés respiratoires pendant le sommeil (apnées) chez près de 10% des patients, une baisse significative des plaquettes sanguines (thrombocytopénie) chez 4%. Ces atteintes semblent plus fréquentes que dans la population générale et nécessitent une prise en charge médicale (assistance respiratoire ou médicaments).
- Mori-Yoshimura M et al. Muscle Nerve . 2021 Oct ;
- Yoshioka W et al. 8 Clin Neurol Neurosurg. 2021 Nov ;
- Guo X et al. Neurol Sci . 2022 Apr
Dans la myopathie distale liée à AN05
Une analyse en IRM des muscles de 17 personnes atteintes de myopathie liée à ANO5 montre un remplacement du tissu musculaire par de la graisse dans les muscles ischio-jambiers et adducteurs des cuisses et du mollet. Les muscles du dos sont relativement préservés. L’atteinte musculaire semble moins importante chez les femmes que chez les hommes.
Dans la myopathie distale liée au gène VCP
Une étude internationale publiée en juillet 2022 avec 255 participants montre l’histoire naturelle de cette myopathie (ou myopathie à inclusions associée à une maladie de Paget osseuse et à une démence fronto-temporale) : 50% des personnes présente une faiblesse musculaire généralisée, 40% un degré d’insuffisance respiratoire, 38% une maladie osseuse de Paget, 21% de la dysautonomie et 14% une démence fronto-temporale.